%202%20naranjas-01.png)
Cri-du-chat: El Síndrome del Maullido del Gato
- Juan Pablo Dovarganes Queipo

- Aug 18, 2025
- 6 min read
Updated: Apr 28

Nanolab te informa sobre el síndrome de cri-du-chat, una enfermedad que provoca que los niños emitan sonidos similares a los maullidos de un gato. De ahí su nombre francés que significa “grito de gato”. Este síndrome tiene un origen genético fascinante: ocurre cuando se pierde un poco de información genética en el cromosoma 5. Acompáñanos a descubrir qué es el síndrome de cri-du-chat, cómo se relaciona con las microdeleciones cromosómicas, y cuáles son las opciones para su prevención y diagnóstico.
Microdeleciones cromosómicas y el síndrome de cri-du-chat
El síndrome de cri-du-chat es el resultado de una microdeleción cromosómica, que ocurre cuando se pierde un fragmento de un cromosoma. Para comprenderlo mejor, analicemos la estructura de los cromosomas. Estos son moléculas de ADN altamente compactadas, lo que permite almacenar una gran cantidad de información en un espacio diminuto, como una célula. Cada especie tiene un número diferente de cromosomas. Esto no determina su complejidad: ¡tener más o menos cromosomas no la hace superior!.
Normalmente, los humanos tenemos 46 cromosomas: 23 que heredamos de la madre y 23 del padre. Cuando un óvulo y un espermatozoide se fusionan para formar un nuevo individuo, el embrión tendrá 46 cromosomas. Sin embargo, en algunos casos, parte del material genético puede perderse o duplicarse. Dependiendo de la magnitud del cambio, hablamos de aneuploidías o microcambios estructurales.
Tanto las aneuploidías como los microcambios estructurales son cromosomopatías, enfermedades causadas por cambios estructurales en los cromosomas. Las primeras afectan grandes cantidades de información genética, ya que implican la pérdida o repetición de un cromosoma completo. Por ejemplo, el síndrome de Down se debe a la copia extra del cromosoma 21. En cambio, los microcambios afectan solo una región del cromosoma. Existen las microdeleciones (pérdida de genes), que son más comunes, y las microduplicaciones (repetición de genes), que son raras. El síndrome de cri-du-chat se debe a una microdeleción en el cromosoma 5.
¿Por qué ocurren las cromosomopatías?
Tanto las microdeleciones como las aneuploidías surgen por errores durante la división celular. Todas las células de nuestro cuerpo tienen 46 cromosomas. Sin embargo, los espermatozoides y los óvulos deben contener la mitad: 23 cromosomas. De lo contrario, cada generación humana acumularía más cromosomas de los necesarios. Para que las células reproductivas logren esto, pasan por un proceso especial de división celular llamado meiosis.
Las células madre productoras de espermatozoides y óvulos (llamadas células germinales) también poseen 46 cromosomas. Sin embargo, gracias a la meiosis, sus células hijas terminan con solo 23 cromosomas. Durante la meiosis, la información genética se redistribuye entre las células madre e hijas. De una sola célula germinal, se generan cuatro células hijas, cada una con 23 cromosomas. No obstante, al mover tanta información, a veces, ocurren errores: una célula hija puede terminar con 22 o 24 cromosomas o un cromosoma puede perder o ganar material genético. Estos eventos son, en su mayoría, naturales y aleatorios.
El síndrome de cri-du-chat y el cromosoma 5
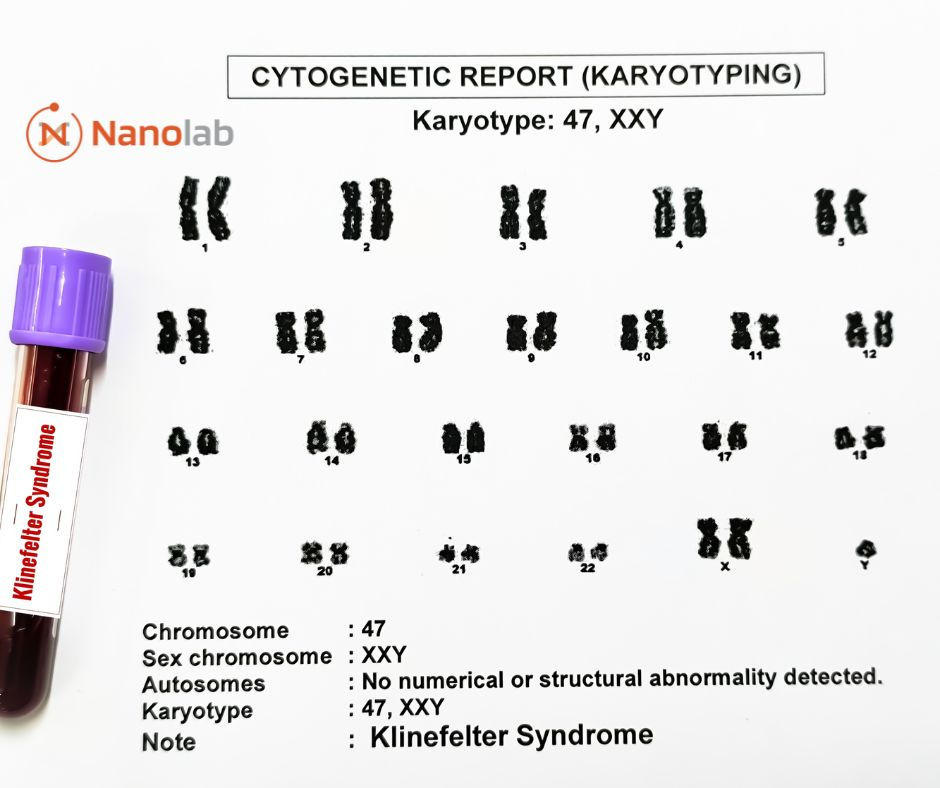
Los cromosomas están organizados en 23 pares. Un cariotipo, la herramienta genética que permite visualizarlos, los ordena por tamaño: el par 1 el más grande y el par 22 el más pequeño. El par 23 está reservado para los cromosomas sexuales: XX en niñas y XY en niños. Cada cromosoma contiene genes específicos y, aunque su ubicación puede parecer aleatoria, cada gen está en un lugar concreto.
El síndrome de cri-du-chat se debe a una microdeleción en el brazo p del cromosoma 5. Para entenderlo mejor, imaginemos la forma clásica de un cromosoma, que parece una letra “X”. Esta forma sugiere cuatro brazos iguales, pero los cromosomas tienen brazos de distinto tamaño: el brazo largo (q), que posee más información genética y el brazo corto (p), que posee menos información. Cuando se menciona una pérdida en el brazo p, significa que se han perdido genes en el brazo corto del cromosoma.
¿Cuántos genes se pierden en el síndrome de cri-du-chat? La cantidad varía, y esto explica por qué cada paciente es único. Las microdeleciones que causan este síndrome ocurren en regiones críticas del síndrome de cri-du-chat, donde la pérdida de ciertos genes están altamente relacionados con el síndrome. Dependiendo de la información genética perdida, los síntomas son más graves o moderados.
Aunque se pueden perder múltiples genes, algunos son especialmente relevantes para este síndrome y se han estudiado con más profundidad. Aquí puedes ver los más importantes:
FLJ25076/UBC-E2: Codifica una proteína que funciona como un sistema de limpieza, eliminando proteínas viejas o dañadas. Su pérdida podría estar relacionada con el sonido característico, similar a un maullido, que producen las personas afectadas.
SEMA5A: Perteneciente a la familia de las semaforinas, es clave para el desarrollo neurológico, ya que guía a las neuronas para que establezcan conexiones.
CTNND2: Facilita la formación de conexiones sinápticas entre neuronas, esenciales para la función cerebral.
TERT: Codifica una telomerasa, proteína que mantiene la estabilidad del cromosoma. Su pérdida podría permitir cambios genéticos adicionales, contribuyendo a la diversidad de síntomas del síndrome.
Síntomas en el síndrome de cri-du-chat
No hay dos pacientes iguales con síndrome de cri-du-chat. Incluso personas con microdeleciones similares pueden presentar síntomas distintos. A continuación, te compartimos algunos de los síntomas más comunes:
Llanto característico: Emiten un sonido similar al maullido de un gato debido a cambios en la laringe. Puedes escucharlo aquí.
Microcefalia: Tamaño de la cabeza más pequeño de lo normal.
Maloclusión dental.
Cara de luna: Rostro redondo y aplanado.
Hipertelorismo: Ojos más separados de lo normal.
Tabique nasal largo.
Personalidad gentil.
Apego a objetos inanimados.
Discapacidad mental moderada a severa.
Comprensión del lenguaje superior a su expresión: Aunque comprenden el lenguaje, no suelen utilizarlo.
Movimientos repetitivos.
Pérdida del color del pelo a edades infantiles.

El diagnóstico para el síndrome de cri-du-chat
La detección del síndrome de cri-du-chat se lleva a cabo utilizando herramientas genéticas especializadas. Ante síntomas sospechosos, el primer paso es un cariotipo para visualizar los cromosomas. Sin embargo, a veces, la microdeleción no es detectable. En estos casos se recurre a un estudio llamado FISH. Esta técnica es similar a un cariotipo, pero utiliza sondas fluorescentes que se unen a ciertos genes específicos, haciendo que el cromosoma brille. La ausencia de fluorescencia, indica la falta de los genes buscados. Aprende más sobre cariotipos aquí.
Durante el embarazo, también es posible evaluar si el bebé presenta este síndrome mediante un cariotipo con amniocentesis, donde se analiza una muestra del líquido amniótico. Este estudio se realiza solo si el médico detecta indicadores específicos en los ultrasonidos.
Otra técnica es la PCR que permite crear muchas copias de ADN para analizar la secuencia genética del brazo p del cromosoma 5 y detectar la ausencia de genes.
El síndrome de cri-du-chat suele ser un evento aleatorio. Por lo que es difícil prevenirlo. No obstante, las parejas que optan por el proceso de fertilización in vitro pueden considerar las pruebas PGT, que analizan el estado genético de los embriones antes de transferirlos al útero. Aprende más sobre las pruebas PGT aquí.
En Nanolab, tu Centro de Genética Integral, realizamos estudios como cariotipos, FISH, PCR y PGT, entre otros estudios genéticos. Además, en Nanolab contarás con el apoyo de un equipo multidisciplinario que incluye médicos, genetistas y psicólogos, que te acompañarán a cada paso el proceso. ¡Conoce más sobre Nanolab CGI y todos nuestros servicios!

Referencias
Ajitkumar A, Jamil RT, Mathai JK. Cri Du Chat Syndrome. [Updated 2022 Oct 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482460/
Espirito Santo, L. D., Moreira, L. M., & Riegel, M. (2016). Cri-Du-Chat Syndrome: Clinical Profile and Chromosomal Microarray Analysis in Six Patients. BioMed research international, 2016, 5467083. https://doi.org/10.1155/2016/5467083
Corrêa, T., Feltes, B. C., & Riegel, M. (2019). Integrated analysis of the critical region 5p15.3-p15.2 associated with cri-du-chat syndrome. Genetics and molecular biology, 42(1 suppl 1), 186–196. https://doi.org/10.1590/1678-4685-GMB-2018-0173
Piovani, G., Ferraro, R. M., & Giliani, S. C. (2025). Establishment and characterization of Cri Du Chat neuronal stem cells: a novel promising resource to study the syndrome. Human cell, 38(4), 98. https://doi.org/10.1007/s13577-025-01230-x
Assendorp, N., Fossati, M., Libé-Philippot, B., Christopoulou, E., Depp, M., Rapone, R., Dingli, F., Loew, D., Vanderhaeghen, P., & Charrier, C. (2024). CTNND2 moderates the pace of synaptic maturation and links human evolution to synaptic neoteny. Cell reports, 43(10), 114797. https://doi.org/10.1016/j.celrep.2024.114797
Przylepa, K., & McKusick, V. (2020). Entry - #123450 - Cri-du-Chat Syndrome - OMIM - (omim.org). OMIM. https://omim.org/entry/123450



Comments